
Alagille syndrome overview: What you need to know
Last updated Feb. 1, 2024, by Marisa Wexler, MS
 Fact-checked by Marta Figueiredo, PhD
Fact-checked by Marta Figueiredo, PhD
Alagille syndrome is a genetic disease that can cause a wide range of symptoms, typically including liver problems as well as heart issues and characteristic changes in the shape of the face. The disorder is also sometimes referred to as Alagille-Watson syndrome or as arteriohepatic dysplasia.
While it’s estimated to affect 1 in every 30,000 newborns, some cases of Alagille syndrome may go undiagnosed, or be misdiagnosed as another condition. This occurs because the disease has potentially overlapping symptoms with other disorders, such as extrahepatic biliary atresia.
The condition affects people of different sexes, races, ethnicities, and geographical regions similarly.
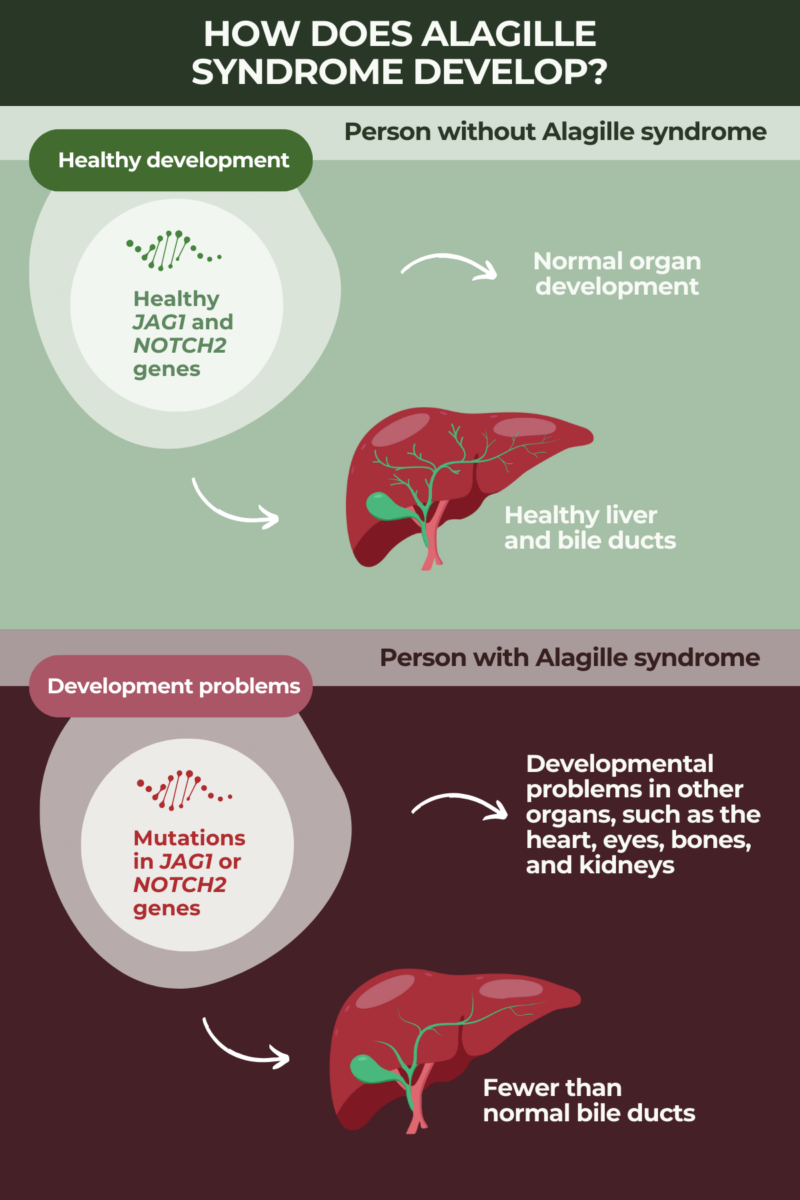
Causes and genetics
Alagille syndrome is a congenital genetic disorder, meaning that the disease is caused by mutations that are present when a person is born. In the vast majority of cases, Alagille syndrome causes are mutations in the JAG1 gene. More rarely, in about 2.5% of cases, the disease is caused by mutations in another gene called NOTCH2.
The JAG1 gene and NOTCH2 gene both belong to the same signaling pathway, called Notch. They provide instructions to produce two different proteins that interact with each other, activating the signaling cascade. Because this signaling is involved in embryonic development, mutations in these genes are thought to lead to abnormalities and errors during early development that ultimately give rise to Alagille syndrome symptoms.
There also are a handful of Alagille syndrome cases in which a specific disease-causing mutation cannot be identified.
Symptoms
Alagille syndrome can cause a wide range of symptoms — and the disease can affect each patient differently. Some individuals experience life-threatening complications, while others may have disease signs that are only noticeable with medical testing. Even among people in the same family with the same disease-causing mutation, Alagille syndrome symptoms can vary.
Besides liver damage, which is one of the disease’s main features, Alagille syndrome may cause abnormalities in the heart, blood vessels, bones, kidneys, and pancreas — each with their own accompanying symptoms. Some patients will experience abnormalities of the facial features, known as Alagille syndrome face, which often includes symptoms affecting the eyes.
Liver problems
Liver problems are one of the most common Alagille syndrome symptoms, which are mostly caused by intrahepatic bile duct paucity, or a reduced number of bile ducts inside the liver. However, other structural bile duct abnormalities also may be the cause.
The bile ducts are a series of tubes that normally carry bile from the liver to the small intestine. Bile is a fluid made by the liver that helps with fatty molecule breakdown during digestion. It also helps with the removal of certain waste products from the body.
In people with bile duct paucity, bile isn’t able to flow normally out of the liver. The slowing or stalling of bile flow, a condition known as cholestasis, causes bile to build up inside the liver, where it ultimately promotes tissue scarring that affects the organ’s function.
In a baby with Alagille syndrome, liver disease typically becomes apparent in the first three months of life, manifesting with signs and symptoms such as yellowed coloring of the skin and whites of the eyes, or jaundice, and increased levels of liver damage markers in the blood.
Liver damage also can result in itchy skin and xanthomas, or raised skin lesions that typically have a waxy, yellowish appearance and are linked to the accumulation of cholesterol deposits underneath the skin. Alagille syndrome patients with liver problems may also have dark urine, intense, pale-colored stools, and an enlarged liver or spleen.
In about 15% of Alagille patients, progressive liver disease leads to liver scarring, also known as cirrhosis, and liver failure, which can be life-threatening and may require a liver transplant.
Alagille syndrome patients also are at increased risk of developing a type of liver cancer called hepatocellular carcinoma.
Heart problems
Heart problems also are a common feature of Alagille syndrome, estimated to affect at least 90% of all cases.
The most frequent issue is heart murmur, or an extra, unusual sound during a heartbeat. This often is due to pulmonary artery stenosis, a structural abnormality in the pulmonary arteries that carry blood from the heart to the lungs. Such congenital heart defects, which are seen in about two-thirds of patients, are marked by a narrowing of these blood vessels, making it harder for the heart to pump blood out.
As many as 16% of children with Alagille syndrome have a complex heart defect called tetralogy of Fallot, which is characterized by four co-occurring abnormalities:
- pulmonary artery stenosis
- ventricular septal defect, or a hole between the lower heart chambers (right and left ventricles)
- enlarged right ventricle
- a displaced aorta, the body’s largest vessel, that causes blood to flow into the aorta from both the right and left ventricles instead of just the left ventricle as is typical.
Characteristic facial features
Most people with Alagille syndrome show characteristic facial features. An Alagille syndrome face, as it is known, includes a broad forehead, deeply set and widely spaced eyes, a straight nose with a bulbous tip, and a pointed chin. These features are said to make the face look like a triangle with the tip pointing down.
One specific eye abnormality, known as posterior embryotoxon, is reported to affect more than three-quarters of people with Alagille syndrome. This condition, which also is present in as much as 15% of the general population, is characterized by a white ring lining the cornea — the thin, transparent membrane that covers the eyeballs. Usually detected via an eye exam, posterior embryotoxon does not typically cause any vision problems, but can help in the diagnosis of Alagille.
Bone abnormalities
People with Alagille syndrome may show certain skeletal abnormalities, the most common being butterfly vertebrae. This condition gets its name due to an irregular shape of the vertebrae, or the bones that make up the spine, which gives these bones a shape that resembles a butterfly.
Butterfly vertebrae usually are visible on an X-ray of the spine, but this skeletal anomaly does not typically cause any noteworthy problems or symptoms for patients.
Craniosynostosis, a condition where the bones in a baby’s skull fuse too early, is more common among Alagille patients than in the general population, though this complication impacts less than 1% of patients.
Individuals with Alagille syndrome may be at increased risk of having broken bones and growth deficiencies due to liver problems that make it harder for the body to absorb needed nutrients.
Diagnosis
Because symptoms of this rare disease often vary greatly from person to person, achieving an Alagille syndrome diagnosis can be challenging. Physicians may suspect Alagille syndrome in people with symptoms and signs suggestive of the disorder and after other potential causes have been ruled out.
A hallmark of the disease, bile duct paucity, can be confirmed with a liver biopsy, in which a small piece of the organ is removed surgically and taken to a laboratory for microscopic study. However, bile duct paucity seems to progress with age, and liver biopsy results can look normal in about a third of patients younger than 6 months of age.
To confirm an Alagille syndrome diagnosis, at least three of the following seven signs and symptoms must be present:
- characteristic facial features
- symptoms of liver disease or cholestasis, which usually is diagnosed via endoscopic imaging
- pulmonary stenosis or other disease-related heart abnormalities, typically detected via imaging scans
- butterfly vertebrae or other disease-typical skeletal abnormalities, detected by X-ray
- posterior embryotoxon or other disease-typical eye abnormalities, detected by a detailed eye examination
- underdeveloped kidneys or other Alagille-related kidney abnormalities, normally detected via an ultrasound
- blood vessel malformations or other vascular abnormalities common to Alagille, detected by a detailed imaging analysis of blood vessels.
Endoscopic imaging typically involves the insertion of an endoscope — a long, flexible tube with an attached light source and tiny video camera — down the mouth and throat, through the stomach, to the first part of the small intestine. This procedure, less invasive than a biopsy, allows the observation of the liver and bile duct system.
An Alagille syndrome diagnosis also can be confirmed by genetic testing to identify a disease-causing mutation in the JAG1 or NOTCH2 genes. In people with such a mutation or a known family history of Alagille syndrome, fewer of the above-listed five characteristics are needed to confirm a clinical diagnosis.
Blood tests also can aid in diagnosing Alagille syndrome by measuring levels of liver enzymes, fatty molecules that usually are broken down by bile acids during digestion, and fat-soluble vitamins, often found at lower levels in these patients.
An early diagnosis of Alagille syndrome is important to ensure proper and timely care. The earlier confirmation of the disease occurs, the sooner treatments can be started.
Treatment
There is not a cure for Alagille syndrome, nor are there any available disease-modifying therapies, or treatments to address the root causes of the condition. Instead, management of Alagille syndrome is focused on treatments that address the specific symptoms and disease manifestations each patient experiences.
A multidisciplinary team of experts often is needed to manage Alagille patients, who should undergo initial examinations of the liver, heart, eyes, kidney, and certain blood vessels. Subsequent regular monitoring of identified abnormalities also should be done.
Patients with cholestasis may be given ursodiol or ursodeoxycholic acid (UDCA), a medication that helps bile to move through the bile ducts. It is sold under the brand names Urso and Actigall.
UDCA may help reduce Alagille syndrome symptoms such as itchy skin, which is thought to be associated with excess bile acids in the bloodstream.
There also are two therapies approved by the U.S. Food and Drug Administration to treat itching in Alagille: Livmarli (maralixibat) and Bylvay (odevixibat). Livmarli is designed for patients ages 3 months and older, while Bylvay is approved for those 1 year and older. Both are taken orally and work to increase the amount of bile acids that are excreted in stool.
Liver dysfunction in Alagille syndrome can make it harder for the body to absorb certain nutrients. Therefore, supplements of some vitamins — most notably vitamins A, D, E, and K — may be given. Similarly, infants with Alagille syndrome may receive a special formula that contains medium chain triglycerides, a type of fatty molecule that’s more easily digested by people with reduced bile flow. In some cases, feeding through a tube connected to the stomach may be needed to ensure patients can get enough nutrition.
Partial biliary diversion, a surgical procedure that involves redirecting the flow of bile, also may be used to manage liver problems in Alagille syndrome, especially if medications and dietary changes are not sufficient.
If liver damage becomes severe, a liver transplant — where a healthy liver from a donor is surgically implanted to replace a patient’s diseased liver — may be required.
Life expectancy
Given that Alagille syndrome is a lifelong condition that can cause fatal complications such as liver failure and heart problems, early interventions and regular monitoring are critical for disease management. Alagille syndrome life expectancy generally is normal for those with mild symptoms, but severe complications are linked to a worse prognosis and reduced lifespan. Still, the long-term outlook for the disease continues to improve as medical science gets better and better.
According to a 2023 study, which assessed outcomes for more than 1,400 Alagille syndrome patients born between 1997 and 2019, more than 85% of children with Alagille syndrome now survive beyond the age of 18.
However, the study also reported that about 30% of patients required a liver transplant during follow-up, with most transplants being needed before adulthood. Other studies have similarly reported that liver transplants are required in about one-third of patients and mostly take place in the first decade of life.
Challenges and coping strategies
Living with Alagille syndrome can present unique challenges, from physical difficulties to emotional and mental health struggles. Although Alagille syndrome cannot be cured, there are therapies that can help manage its symptoms and prevent serious complications. Patients and their families are advised to work with their healthcare team to come up with a personalized strategy to best manage the disease.
Support also is available for people with Alagille syndrome, including from Mirum Pharmaceuticals, the company that markets Livmarli. Mirum offers a website with a range of downloadable resources, including guides to help patients and their families have productive discussions with healthcare providers. Checklists to help caregivers stay on top of everything that needs to be covered, and activities to educate children about Alagille, also are available on the website.
Additionally, a nonprofit called the Alagille Syndrome Alliance (ALGSA) is dedicated to giving support to families affected by the disease. Among other resources, the alliance offers a range of support groups for people affected by Alagille. There are specific groups for men, women, and children, and for those living in certain countries. These groups may help equip patients with coping mechanisms that can assist in the day to day of living with Alagille syndrome.
Liver Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
-

-
 Discussion
Discussion
-

-
-
 Discussion
Discussion